

Laboratory News
20 Years of Crystal Hits
Science

Diffraction-based structural methods contribute a large fraction of the biomolecular structural models available, providing a critical understanding of macromolecular architecture. These methods require crystallization of the target molecule, which remains a primary bottleneck in crystal-based structure determination. The National High-Throughput Crystallization Center at Hauptman–Woodward Medical Research Institute has focused on overcoming obstacles to crystallization through a combination of robotics-enabled high-throughput screening and advanced imaging to increase the success of finding crystallization conditions. This paper will describe the lessons learned from over 20 years of operation of our high-throughput crystallization services. The current experimental pipelines, instrumentation, imaging capabilities and software for image viewing and crystal scoring are detailed. New developments in the field and opportunities for further improvements in biomolecular crystallization are reflected on.
Appointment to International Book Committee
News

Dr. Snell was appointed to the International Union of Crystallography (IUCr) Committee as a member of the Book Series Committee. The Committee recommends new book publications to the IUCr Executive Committee and the Delegates of Oxford University Press. Recommended publications are IUCr monographs, which are in-depth expositions of specialized topics in crystallography, crystallography texts providing standardized methods and approaches, and related books.
Landscape of a Cold Unfolding Protein
Science
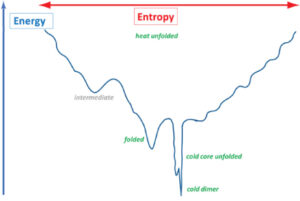
Understanding protein folding is crucial for protein sciences. We created the cold unfolding 4-helix bundle DCUB1 with a de novo designed bipartite hydrophilic/hydrophobic core featuring a hydrogen bond network which extends across the bundle in order to study the relative importance of hydrophobic versus hydrophilic protein–water interactions for cold unfolding. Structural and thermodynamic characterization resulted in the discovery of a complex energy landscape for cold transitions, while the heat unfolded state is a random coil. Our results suggest that cold unfolding is initiated by the intrusion of water into the hydrophilic core network and that cooperativity can be tuned by varying the number of core hydrogen bond networks. Protein design has proven to be invaluable to explore the energy landscapes of cold states and to robustly test related theories.
COVID-19 Research
Science
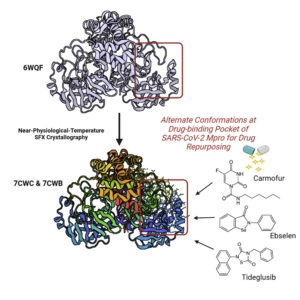
The COVID-19 pandemic has resulted in 198 million reported infections and more than 4 million deaths as of July 2021. To assist in drug repurposing and design, we determined two apo structures of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease at ambient temperature by serial femtosecond X-ray crystallography. We employ detailed molecular simulations of selected known main protease inhibitors with the structures and compare binding modes and energies. The combined structural and molecular modeling studies not only reveal the dynamics of small molecules targeting the main protease but also provide invaluable opportunities for drug repurposing and structure-based drug design strategies against SARS-CoV-2.
SAXS studies of radical scavengers
Science

X-ray-based techniques are a powerful tool in structural biology but the radiation-induced chemistry that results can be detrimental and may mask an accurate structural understanding. Using a protein designed to undergo a large-scale structural change from breakage of a disulfide bond, radiation damage can be monitored with small-angle X-ray scattering. We have quantitatively evaluated how three scavengers commonly used in crystallographic experiments – sodium nitrate, cysteine, and ascorbic acid – perform in solution at 10°C. The SAXS-based method can detect damage at X-ray doses far lower than those accessible crystallographically, thereby providing a detailed picture of scavenger processes. Our engineered approach might provide a platform for more systematic and comprehensive screening of radioprotectants that can directly inform mitigation strategies for both solution and crystallographic experiments, while also clarifying fundamental radiation damage mechanisms and allowing the study of low dose radiation.
Engineering High-Resolution Insight
Science
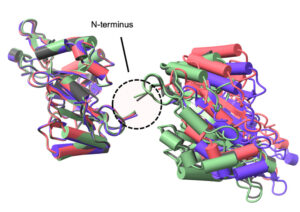
Radition chemistry in biological X-ray crystallography iproduces both global and site-specific damage. Site specific damage can misdirect the biological interpretation of the structural models produced. The doses used for X-ray crystallography are typically in the kilo-gray to mega-gray range. While disulfide bonds are among the most significantly affected species in proteins in the crystalline state at both cryogenic and higher temperatures, there is limited information on their response to low X-ray doses in solution, the details of which might inform biomedical applications of X-rays. An engineered a protein approach enables small angle X-ray scattering (SAXS), to detect and monitor a residue specific process. The ability to generate a large-scale change through a site-specific interaction represents a promising tool for advancing radiation damage studies under solution conditions and in dose regimes of health-related impact.
Structural Biology Fighting COVID-19
Science
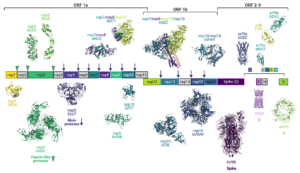
The global COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) wreaked unprecedented havoc on global society. Yet the sheer magnitude and uniqueness of this event has also spawned a massive mobilization of effort in the scientific community to investigate the virus, to develop therapeutics and vaccines, and to understand the public health impacts. Structural biology has been at the center of these efforts, and we reflect on the status of structural science vis-a`-vis its role in the fight against COVID-19, to register the unprecedented response and to contemplate the role of structural biology in addressing future outbreak threats. Examining SARS-CoV-2 protein structures that have been deposited in the PDB since the virus was first identified allows a unique window into the power of structural biology and a snapshot of the advantages of the different techniques available, as well as insight into the complementarity of the structural methods
Elected to ACA Fellow
News

Dr. Edward Snell was elected as American Crystallographic Association Fellow. The American Crystallographic Association – the Structural Science Society – established the Fellows distinction to recognize a high level of excellence in scientific research, teaching, and professional duties, but also service, leadership, and personal engagement in the world of crystallography and science.
Crystallization in Space
Science

More regular space flight is becoming a reality, new diffraction data detectors have become available that have both a faster readout and lower noise, a new generation of extremely bright X-ray sources and X-ray free-electron lasers (XFELs) have become available with beam collimation properties well suited geometrically to more perfect protein crystals. Neutron sources, instrumentation, and methods have also advanced greatly for yielding complete structures at room temperature and radiation damage-free. The larger volumes of protein crystals from microgravity can synergise well with these recent neutron developments. Unfortunately, progress in harnessing these new technologies to maximize the benefits seen in microgravity-grown crystals has been patchy and even disappointing. Despite detailed theoretical analysis and key empirical studies, crystallization in microgravity has not yet produced the results that demonstrate its potential. In this review we present some of the key lessons learned and show how processes could yet be optimized given these new developments
Networks Across Scales
Science
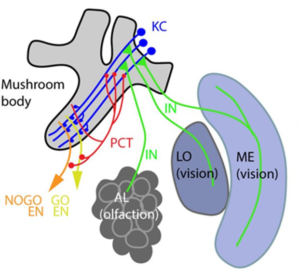
A wide range of networks exist in biology, from gene regulatory networks important for organismal development, protein interaction networks that govern physiology and metabolism, and neural networks that store and convey information to networks of microbes that form microbiomes within hosts, animal contact networks that underlie social systems, and networks of populations on the landscape connected by migration. Increasing availability of extensive (big) data is amplifying our ability to quantify biological networks. Similarly, theoretical methods that describe network structure and dynamics are being developed. In this paper, we explore the opportunity for discovering universal laws connecting the structure of biological networks with their function, positioning them on the spectrum of time-evolving network structure, that is, dynamics of networks, from highly stable to exquisitely sensitive to perturbation. If such general laws exist, they could transform our ability to predict the response of biological systems to perturbations—an increasingly urgent priority in the face of anthropogenic changes to the environment that affect life across the gamut of organizational scales.
Structures involving the laboratory
X-ray radiation study on xylose isomerase
6QRY
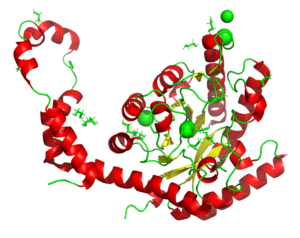
A radiation chemistry study on the impacr of dose on structural information using xylose isomerase as an example. The results show that information interpreted as mechanism can be a result of the X-rays used to produce the models. See also PDB entries 6QRX, 6QRW, 6QRV, 6QRU, 6QRT, 6QRS, 6QRR.
Latency Associated Peptide unbound to TGF-β1
6P7J
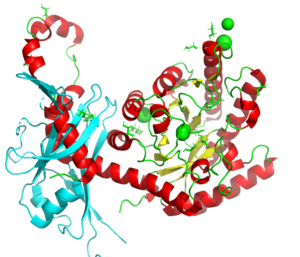
A study on the dissociation of the inhibitory protein latency associated peptide (LAP) from transforming growth factor β1, important for those undergoing radiation therapy.
The C-terminal domain (277-440) of a putative chitobiase from Bacteroides thetaiotaomicron.
6OE2
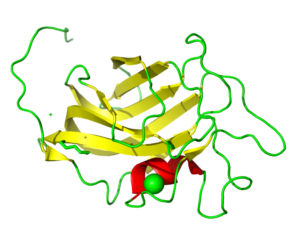
PIXE data revealed Mn as the experimentally determined metal rather than Zn as originally modeled. This changed the coordination number of the metal from 4 to 5. This may have an importance in understanding the basics of Tay-Sachs and Sandhoff disease.
The nucleotide-binding protein AF_226 in complex with ADP from Archaeoglobus fulgidus
6OBY
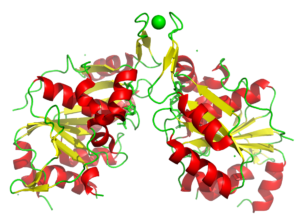
Nucleotide binding proteins play a crucial role in many diseases. In this case PIXE identified Co bound rather than the modeled Zn. It is not known if the Co was promisciously bound and if it is biologically relevant.
A putative histidinol phosphatase hisK from Listeria monocytogenes
6NLR
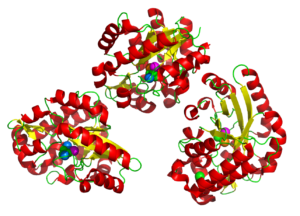
PIXE combined with tha anomalous X-ray signal allowed the metals in the tri-nuclear metal center to be correctly identified replacing the Fe and Zn with Fe, Co, and Mn. A sulfate ion was revealed in the active site agreeing well with literature on related proteins.
Wild type T4 lysozyme structure
4SOW
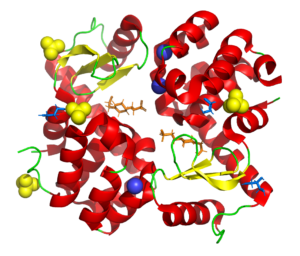
Part of a study aimed at deliberatly de-crystallizing T4 lysozyme using surface mutations to understand the influence of different chemistries on the crystallization process.
Understanding the radiation chemistry around disulfide bond breakage
4H9I
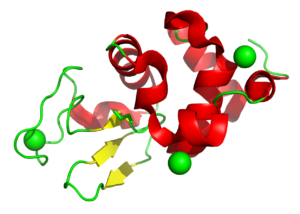
Disulfide bond damage is seen in X-ray structures but is also caused by free radical production in the body and drives key biological mechanisms. This study used lysozyme to study this process. Related models are 4H9E, 4H9C, 4H9B, 4H9A, 4H94, 4H93, 4H8Z, 4H8Y, and 4H8X.
The Structure of the Pand/Panz Protein Complex
4CS0
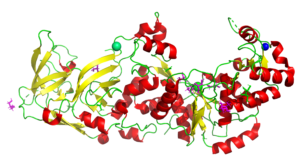
In this study our collaborators determined the structure and we used SAXS to model the protein complex. The information provides a mechanistic understanding of regulation in the body. See also PDB entries 4CRZ and 4CRY.
The tRNA Binding Domain of Glutaminyl-tRNA Synthetase from Saccharomyces cerevisiae
3TL4
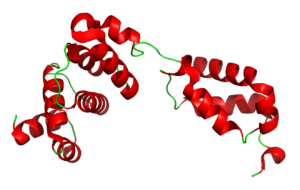
Eukaryotic glutaminyl-tRNA synthetase (GlnRS) contains an appended N-terminal domain (NTD) whose precise function is unknown. The structure of an N-terminal domain enabled a model of the full-length enzyme in solution, and modeling its binding to tRNA suggesting that in eukaryotes there is motion of the C-terminal domain through conserved hinge. The domain is nearly ubiquitious among eukaryotic GlnRS species but lacking in prokaryotic holologs.
Glutaminyl-tRNA Synthetase from Saccharomyces Cerevisiae
4H3S
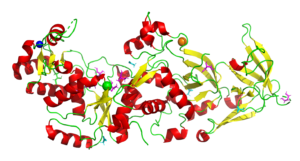
In all organisms, aminoacyl tRNA synthetases covalently attach amino acids to their cognate tRNAs. Many eukaryotic tRNA synthetases have acquired appended domains, whose origin, structure and function are poorly understood. The N-terminal appended domain (NTD) of glutaminyl-tRNA synthetase (GlnRS) is intriguing since GlnRS is primarily a eukaryotic enzyme, whereas in other kingdoms Gln-tRNAGln is primarily synthesized by first forming Glu-tRNAGln, followed by conversion to Gln-tRNAGln by a tRNA-dependent amidotransferase. . Our results suggest a possible origin and function of the NTD that would link the phylogenetically diverse mechanisms of Gln-tRNAGln synthesis.